Маски для быстрого роста волос
24 июля 2023 657 0Длинные, густые и шелковистые волосы всегда были и есть эталоном женской красоты. О них писали в стихах и слагали легенды. Если ваша мечта иметь роскошные волосы,...
Читать далее »
Как увеличить глаза с помощью макияжа
20 июля 2023 741 0Макияж – это создание гармоничного лица, в котором акцент делается на глаза либо на губы. Выделение того и другого в дневном make up считается вульгарным и...
Читать далее »
Свои секреты красоты
18 ноября 2022 809 0Краски для волос аллергику Перед окрашиванием бровей и ресниц вам следует знать о некоторых вещах. Проводить окраску можно только в том случае, если нет...
Читать далее »
Ребёнку психологически комфортно
17 ноября 2022 6916 0Ребёнка вновь интересует грудь мамы Довольно сложная ситуация. Общение ребёнка с грудью матери всегда зеркально показывает взаимопонимание ребёнка и...
Читать далее »
Маму волнуют вопросы
16 ноября 2022 1025 0Почему ребёнок плачет? Плач — своеобразный язык младенцев. Таким образом малыш подаёт нам сигнал о том, что ему, что — то не нравится, он чем — то обеспокоен....
Читать далее »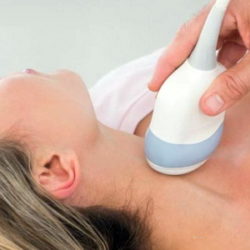
Влияние щитовидной железы и эрозии на роды
14 ноября 2022 525 0Проблемы со щитовидной железой влияют на репродуктивную функцию Заболевания щитовидной железы относятся к экстрагенитальной патологии, которая может...
Читать далее »
Секреты красоты лица
13 ноября 2022 639 0О лазерной чистке лица Лазерная чистка лица проводится при воздействии лазерного луча на проблемные зоны кожи. При таком воздействии ороговевшие клетки...
Читать далее »
Лечение эндометриоза
12 ноября 2022 625 0Эндометриоз Эндометриоз — это состояние, когда по причине травмы, заброса менструальной крови через трубы в брюшную полость, кесарева сечения и т. п.,...
Читать далее »
Агрессивное поведение детей
11 ноября 2022 672 0Борьба малыша за свои права Под агрессией понимается активность детей, и в ней нет ничего плохого. Ваш сын борется за своё право обладать жизненными благами. В...
Читать далее »
С удовольствием кормить малыша грудью
10 ноября 2022 636 0Обстановка при кормлении Во время кормления очень важно уделять ребенку всё ваше внимание (особенно первые 20 минут) — смотреть ему в глазки, делать массажик...
Читать далее »- Готовимся к лету: диеты
- Диеты (видео)
- Женская диета
- Женская красота
- Женская мода
- Женская психология (видео)
- Женские рецепты
- Интимные отношения (видео)
- Красота женщины: уход за собой
- Лучшие книги для женщин
- Любящая мама
- Мода и красота (видео)
- Поздравление женщине
- Психология женщин в отношениях
- Психология женщины
- Разное — полезное
- Самые успешные женщины
- Сохраним здоровье